Bismark工具介绍
简介
Bismark是一个灵活的工具,可以高效地分析BS-Seq数据,方便地进行读段比对和甲基化探测,Bismark能区分CpG、CHG和CHH,允许用户通过可视化来解释数据,首先介绍一下Bismark的比对原理。
亚硫酸氢盐文库有两种不同的类型,第一种情况是以带有方向的方式生成的序列文库,比如实际测序reads将对应于亚硫酸氢盐转换后的原始正向或反向链。在第二种情况下,链的特异性不被保留,这意味着所有四种可能的亚硫酸氢盐DNA链以大致相同的频率进行测序。
由于亚硫酸氢盐处理的reads对应的链特异性是未知的,Bismark旨在通过同时进行四个比对过程来确定唯一比对。首先,亚硫酸氢盐处理的reads进行C到T和G到A的转换。然后,每个reads与进行相同转换的参考基因组通过Bowtie进行比对。如下图所示:
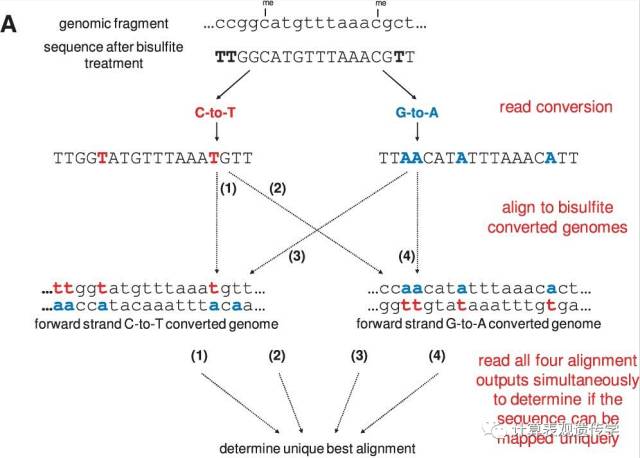
这种比对方式使Bismark可以识别到唯一的reads原始链。 所以,Bismark可以处理来自定向和非定向的BS-Seq数据。由于reads中剩余的C也进行了转换,所以可以准确地处理部分甲基化的情况。
许多以前的BS-Seq程序仅仅用于比对,这意味着获取潜在的甲基化数据需要大量后期处理和计算知识。除了比对功能,Bismark还能确定每个胞嘧啶甲基化状态,如下图所示:
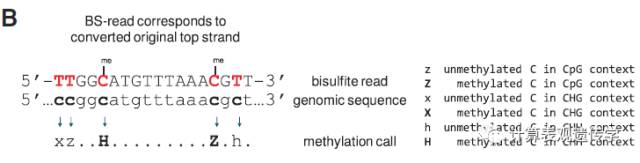
哺乳动物甲基化主要是CpG的形式;然而,但还有一些非CpG的甲基化在胚胎干细胞中。在植物中,甲基化的形式有CpG、CHG、CHH(H可以是A,T或C)。为了分析不同形式的甲基化或模式生物,Bismark考虑了周围环境的序列,并能够区分CpG、CHG、CHH。
Bismark的输出结果中每条read占一行,并包含比对位置,比对的链,亚硫酸氢盐read序列,其比对到的基因组序列。该输出可以进行后续处理或用于提取单个胞嘧啶的甲基化信息。这种甲基化状态的结果由Bismark附带的甲基化提取器产生。该甲基化输出能区分CpG、CHG或CHH,链特异或混合链的格式都可以获得。前者可用于研究不对称甲基化(比如CHH甲基化)。甲基化提取器的输出将为每个胞嘧啶分配一行,使用链信息来编码其甲基化状态:'+'表示甲基,' - '非甲基化。该输出可以转换为其他比对格式,如SAM / BAM,或导入基因组浏览器的SeqMonk,这样就可以被可视化和进一步研究且不需要额外的计算处理。
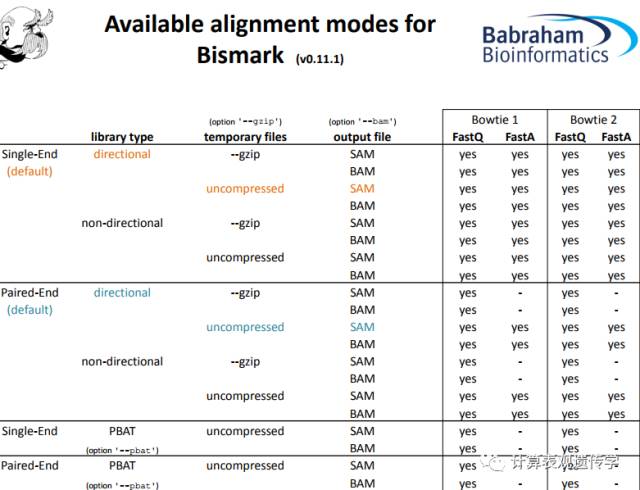
可用比对模式表:
1 Bismark的安装
Bismark工作环境要求:
1,已安装perl和Bowtie(或Bowtie2)
2,需要有参考基因组序列(Bismark支持fasta和fa格式的基因组)
硬件要求:至少5个CpU和12个G的RAM(内存).运行bismark之前最好使用free检查一下内存,大于16G时更不容易出错。
Bismark安装:
从http://www.bioinformatics.babraham.ac.uk/projects/bismark/上下载Bismark,其格式为.tar.gz,在linux系统下解压命令为tar -xzffile.tar.gz。
1 Bismark的使用
1,下载参考基因组:
ftp://ftp.ncbi.nih.gov/genomes/或者http://www.ensembl.org/info/data/ftp/index.html/
2,获取BS-Seq reads数据:
Bismark要求reads为.fa或者.fastq文件(从NCBI上下载的.sra文件需转为.fastq文件,可以使用sratoolkit工具)
3,导入参考基因组,建立比对使用的参考基因组:
命令:bismark_genome_preparation [options] <path_to_genome_folder>
结果:在基因组目录下产生Bisulfite_Genome目录
实例:bismark_genome_preparation --path_to_bowtie/Software/bowtie2-2.0.0-beta7/ --verbose --bowtie2 /Ensemble
参数:
--yes 如果有安全类问题则自动选择yes,比如覆盖某个已存在的文件。
--path_to_bowtie后面跟着的/Software/bowtie2-2.0.0-beta7/为bowtie目录的路径,
--verbose表示输出详细信息以方便调试。
--bowtie2指明此处使用的是Bowtie2,Bowtie1同理。
/Ensemble为需要导入的参考基因组的路径。
注:对于一套数据只需建立一次参考基因组。
4,使用bismark进行比对(使用默认参数):
Bismark支持:
a)reads可以是序列格式:fastq或者fasta,也可以是压缩文件.gz
b)双末端或者单末端
c)read长度可变
d)定向和非定向的BS-Seqlibraries
命令:USAGE: bismark [options] <genome_folder> {-1 <mates1> -2<mates2> | <singles>}
结果:产生两个文件
①文件名_bismark.sam(序列比对结果的详细信息)
②文件名_bismark_mapping_report.txt(比对结果的总结)
实例: bismark -q --phred64-quals-n 1 -l 40 /data/genomes/homo_sapiens/GRCh37/ s_1_sequence.txt
参数:
-q 输入文件为FastQ,文件后缀通常为.fq 或者.fastq。
--phred64-quals 指定FastQ文件的质量分数格式,默认为phred33。
-n seed(read的一部分)中允许的最大错配数,可取0-3,默认为2。
-l 设置seed长度,质量好的一端的reads的碱基数,默认28。
5, 提取甲基化水平
命令:methylation_extractor [options] <filenames>
实例: bismark_methylation_extractor -s -comprehensive --bedGraph --counts--cytosine_report --genome_folder /Ensemble /result/s.fastq_bt2_bismark.sam
更简洁的写法:bismark_methylation_extractor -s -comprehensive --bedGraph --countss.sam
参数:
-s指单末端数据,-comprehensive指把四条链的结果合并为一个文件
--bedGraph指将产生一个BedGraph文件存储CpG的甲基化信息
--counts指在bedGraph中有每个C上甲基化reads和非甲基化reads的数目
--genome_folder后跟着参考基因组的位置
--cytosine_report指报道全基因组所有的CpG。只有当指定
--cytosine_report时才需要genome_folder
(CpG、CHH、CHG的三个文件默认存储在当前目录下,bedGraph文件默认存储在SAM所在的目录下。sam文件和输出目录不能同时为绝对路径)
更详细说明的参见Bismark_User_Guide:
http://www.bioinformatics.babraham.ac.uk/projects/bismark/
参考文献:
Chen,P.Y. et al. (2010) BS Seeker: precisemapping for bisulfite sequencing. BMC
Bioinformatics, 11, 203.
Cokus,S.J. et al. (2008) Bismark: aflexible aligner and methylation caller for Bisulfite-Seq
applications. Bioinformatics Group.
Feng,S. et al. (2010) Conservation anddivergence of methylation patterning in plants
and animals. Proc. Natl Acad. Sci. USA,107, 8689–8694.
Harris,R.A. et al. (2010) Comparison ofsequencing-based methods to profile DNA
methylation and identification ofmonoallelic epigenetic modifications. Nat.
Biotechnol., 28, 1097–1105.
Langmead,B. et al. (2009) Ultrafast andmemory-efficient alignment of short DNA
sequences to the human genome. GenomeBiol., 10, R25.
©著作权归作者所有:来自51CTO博客作者mob604756f4ef89的原创作品,如需转载,请注明出处,否则将追究法律责任