单细胞分析实录(19): 基于CellPhoneDB的细胞通讯分析及可视化 (下篇)
单细胞分析实录(19): 基于CellPhoneDB的细胞通讯分析及可视化 (下篇)
在上一篇帖子中,我介绍了CellPhoneDB的原理、实际操作,以及一些值得注意的地方。这一篇继续细胞通讯分析的可视化。
公众号后台回复20210723获取本次演示的测试数据,以及主要的可视化代码。
所有的数据和结果文件均已打包,下载后直接就能跑下面的代码画图。
下面的代码可以绘制对称热图
(如果你不清楚为啥热图要沿着对角线对称,可以看一下之前的推文)
library(tidyverse)
library(RColorBrewer)
library(scales)
pvalues=read.table("./test/pvalues.txt",header = T,sep = "\t",stringsAsFactors = F)
pvalues=pvalues[,12:dim(pvalues)[2]]
statdf=as.data.frame(colSums(pvalues < 0.05))
colnames(statdf)=c("number")
statdf$indexb=str_replace(rownames(statdf),"^.*\\.","")
statdf$indexa=str_replace(rownames(statdf),"\\..*$","")
statdf$total_number=0for (i in 1:dim(statdf)[1]) {
tmp_indexb=statdf[i,"indexb"]
tmp_indexa=statdf[i,"indexa"] if (tmp_indexa == tmp_indexb) {
statdf[i,"total_number"] = statdf[i,"number"]
} else {
statdf[i,"total_number"] = statdf[statdf$indexb==tmp_indexb & statdf$indexa==tmp_indexa,"number"]+
statdf[statdf$indexa==tmp_indexb & statdf$indexb==tmp_indexa,"number"]
}
}
rankname=sort(unique(statdf$indexa))
statdf$indexa=factor(statdf$indexa,levels = rankname)
statdf$indexb=factor(statdf$indexb,levels = rankname)
statdf%>%ggplot(aes(x=indexa,y=indexb,fill=total_number))+geom_tile(color="white")+
scale_fill_gradientn(colours = c("#4393C3","#ffdbba","#B2182B"),limits=c(0,20))+
scale_x_discrete("cluster 1")+
scale_y_discrete("cluster 2")+
theme_minimal()+
theme(
axis.text.x.bottom = element_text(hjust = 1, vjust = NULL, angle = 45),
panel.grid = element_blank()
)
ggsave(filename = "interaction.num.2.pdf",device = "pdf",width = 12,height = 10,units = c("cm"))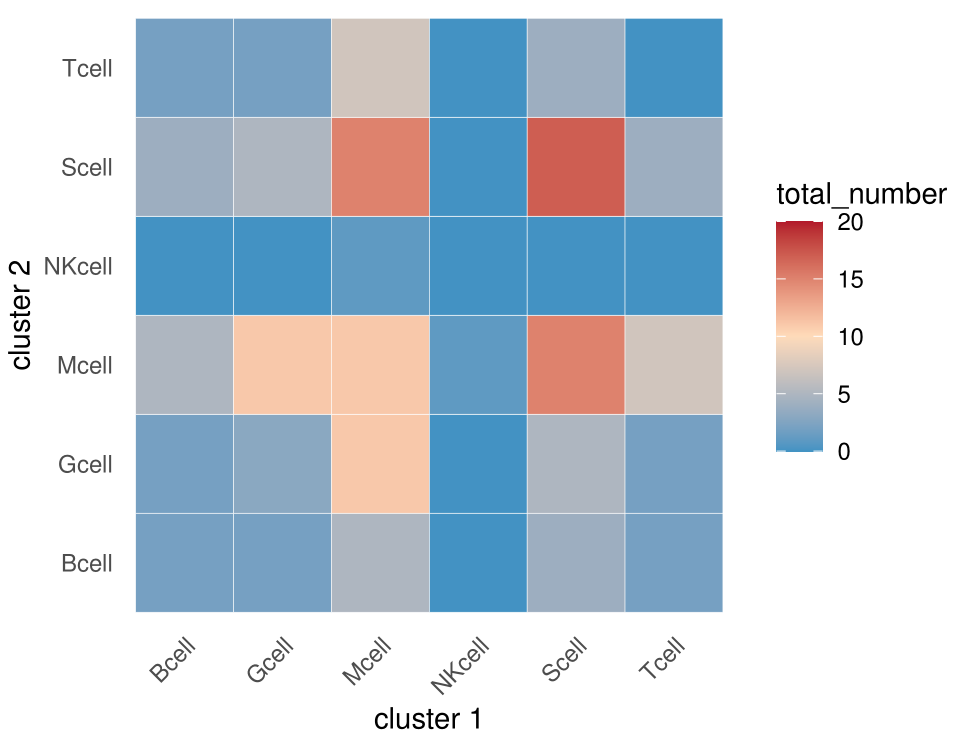
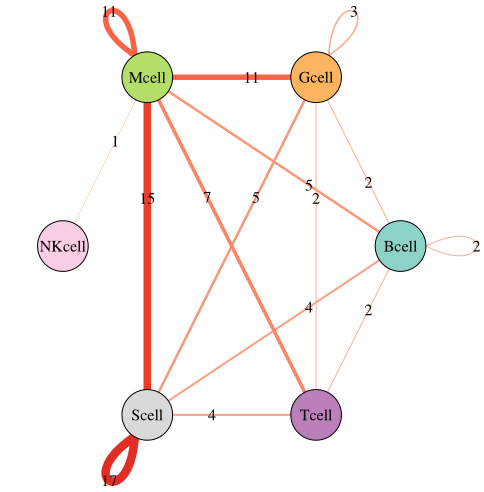
还可以用网络图表示互作关系的数量
代码如下
library(tidyverse)
library(RColorBrewer)
library(scales)
library(igraph)
pvalues=read.table("./test/pvalues.txt",header = T,sep = "\t",stringsAsFactors = F)
pvalues=pvalues[,12:dim(pvalues)[2]]
statdf=as.data.frame(colSums(pvalues < 0.05))
colnames(statdf)=c("number")
statdf$indexb=str_replace(rownames(statdf),"^.*\\.","")
statdf$indexa=str_replace(rownames(statdf),"\\..*$","")
rankname=sort(unique(statdf$indexa))
A=c()
B=c()
C=c()
remaining=ranknamefor (i in rankname[-6]) {
remaining=setdiff(remaining,i) for (j in remaining) {
count=statdf[statdf$indexa == i & statdf$indexb == j,"number"]+
statdf[statdf$indexb == i & statdf$indexa == j,"number"]
A=append(A,i)
B=append(B,j)
C=append(C,count)
}
}
statdf2=data.frame(indexa=A,indexb=B,number=C)
statdf2=statdf2 %>% rbind(statdf[statdf$indexa==statdf$indexb,c("indexa","indexb","number")])
statdf2=statdf2[statdf2$number > 0,] #过滤掉值为0的观测#设置节点和连线的颜色color1=c("#8DD3C7", "#FDB462", "#B3DE69", "#FCCDE5", "#D9D9D9", "#BC80BD")
names(color1)=rankname
color2=colorRampPalette(brewer.pal(9, "Reds")[3:7])(20) #将颜色分成多少份,取决于互作关系数目的最大值names(color2)=1:20 #每一份颜色用对应的数字命名#做网络图##下面的四行代码相对固定net <- graph_from_data_frame(statdf2[,c("indexa","indexb","number")])
edge.start <- igraph::ends(net, es=igraph::E(net), names=FALSE)
group <- cluster_optimal(net)
coords <- layout_in_circle(net, order = order(membership(group)))
E(net)$width <- E(net)$number / 2 #将数值映射到连线的宽度,有时还需要微调,这里除以2就是这个目的E(net)$color <- color2[as.character(ifelse(E(net)$number > 20,20,E(net)$number))] #用前面设置好的颜色赋给连线,颜色深浅对应数值大小E(net)$label = E(net)$number #连线的标注E(net)$label.color <- "black" #连线标注的颜色V(net)$label.color <- "black" #节点标注的颜色V(net)$color <- color1[names(V(net))] #节点的填充颜色,前面已经设置了;V(net)返回节点信息#调整节点位置的线条角度##如果没有这两行代码,节点位置的圆圈是向右的loop.angle<-ifelse(coords[igraph::V(net),1]>0,-atan(coords[igraph::V(net),2]/coords[igraph::V(net),1]),pi-atan(coords[igraph::V(net),2]/coords[igraph::V(net),1]))
igraph::E(net)$loop.angle[which(edge.start[,2]==edge.start[,1])] <- loop.angle[edge.start[which(edge.start[,2]==edge.start[,1]),1]]#pdf("interaction.num.3.pdf",width = 6,height = 6)plot(net,
edge.arrow.size = 0, #连线不带箭头
edge.curved = 0, #连线不弯曲
vertex.frame.color = "black", #节点外框颜色
layout = coords,
vertex.label.cex = 1, #节点标注字体大小
vertex.size = 30) #节点大小#dev.off()气泡图——具体的互作关系
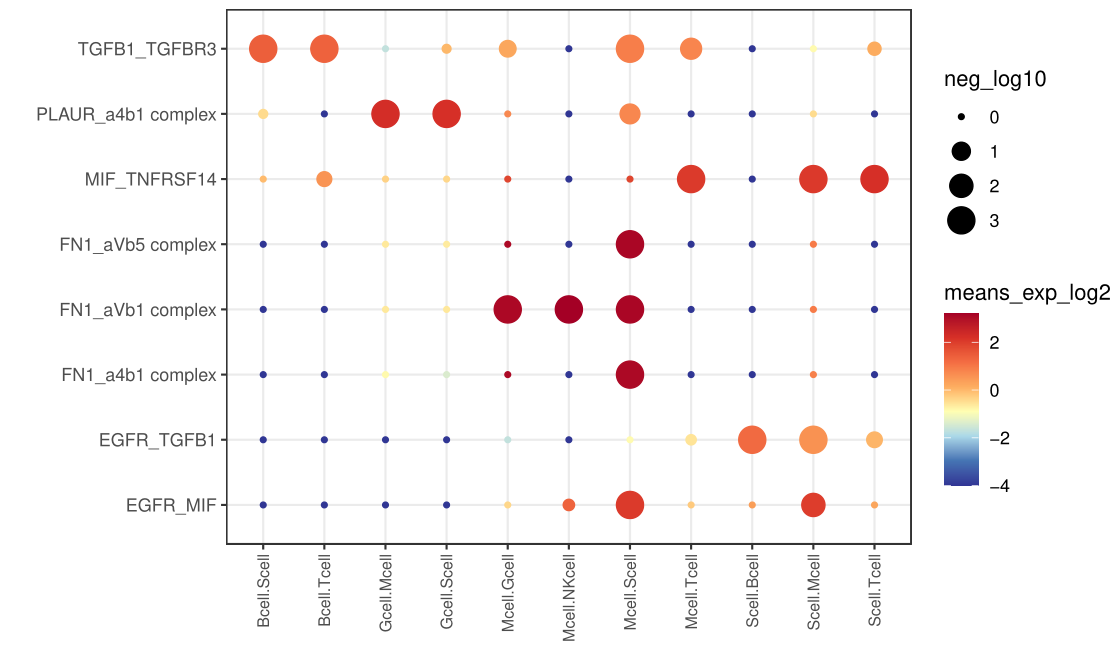
以上几种图,只是用来展示数量,具体的两种细胞之间的互作关系可以用如下的代码展示:
source("CCC.bubble.R")
CCC( pfile="./test/pvalues.txt",
mfile="./test/means.txt",
#neg_log10_th= -log10(0.05),
#means_exp_log2_th=1,
#neg_log10_th2=3,
#means_exp_log2_th2=c(-4,6),
#notused.cell=c("Bcell","Gcell"),
#used.cell=c("Mcell"),
#cell.pair=c("Mcell.Scell","Mcell.NKcell","Mcell.Tcell","Scell.Mcell","NKcell.Mcell","Tcell.Mcell"),#这里是自定义的顺序,若是可选细胞对的子集,则只展示子集,若有交集则只展示交集;空值情况下,会根据可选细胞对自动排序
#gene.pair=c("MIF_TNFRSF14","FN1_aVb1 complex","EGFR_MIF")#作用同上
)
ggsave(filename = "interaction.detail.1.pdf",device = "pdf",width =20,height = 12,units = "cm")参数解释:
neg_log10_th和means_exp_log2_th两个参数用来筛选显著的互作关系;neg_log10_th2和means_exp_log2_th2两个参数用来限定最终气泡图的数值范围;notused.cell不包含的细胞类型;used.cell必须包含的细胞类型;cell.pair必须包含的细胞pair,以及它们的顺序;gene.pair必须包含的基因pair,以及它们的顺序。
后面四个参数在细化气泡图的时候,很有用。
我们先不加额外的参数,查看全部的互作关系
随后再细化,指定需要展示的细胞类型和gene pair,如下:
CCC( pfile="./test/pvalues.txt",
mfile="./test/means.txt",
cell.pair=c("Mcell.Scell","Mcell.NKcell","Mcell.Tcell","Scell.Mcell","NKcell.Mcell","Tcell.Mcell"),#这里是自定义的顺序,若是可选细胞对的子集,则只展示子集,若有交集则只展示交集;空值情况下,会根据可选细胞对自动排序
gene.pair=c("MIF_TNFRSF14","FN1_aVb1 complex","EGFR_MIF")#作用同上
)
ggsave(filename = "interaction.detail.2.pdf",device = "pdf",width =16,height = 10,units = "cm")
最后那个CCC( )函数是小编写的,小编觉得还挺好用的。并不复杂,也才120行。如果你也想用,欢迎转发上一篇推文,截图后发给公众号后台,留下邮箱,小编就会发给你哦。别怪小编套路呀,写这两篇帖子花了不少时间呢
因水平有限,有错误的地方,欢迎批评指正!
来源https://www.cnblogs.com/TOP-Bio/p/15057637.html